Best poster prizes at ‘In situ structural biology: expanding the toolbox for structural cell biology’
The EMBO Workshop ‘In situ structural biology: expanding the toolbox for structural cell biology‘ took place last month at EMBL Heidelberg and virtually.
From cryo-EM to multi-scale modelling – what types of biological questions can be addressed using in situ structural biology? This conference series brings together experts and early stage researchers from different fields and disciplines to define what types of biological questions can already be addressed using in situ structural biology and which might be approached in the future. It highlights recent methodological developments and promotes their usage as new tools for cell biology across scales: from molecules to organelles, cells and, cell-cell interactions. #EMBOinsitu provides a discussion platform for structural biology methods within the cellular context and stimulates conversations about how to meaningfully integrate data from different methods.
For this year’s edition of the conference, we had 211 people attending on-site and 76 participants attending remotely. There were 8 fellowships provided by the EMBL Corporate Partnership Programme and EMBO. With the total of 100 posters to view, we held two poster sessions during which the presenters could discuss their research — their work was then voted for by all participants. There were five poster prizes awarded during the meeting. We are pleased to share with you all five of the winners’ abstracts!
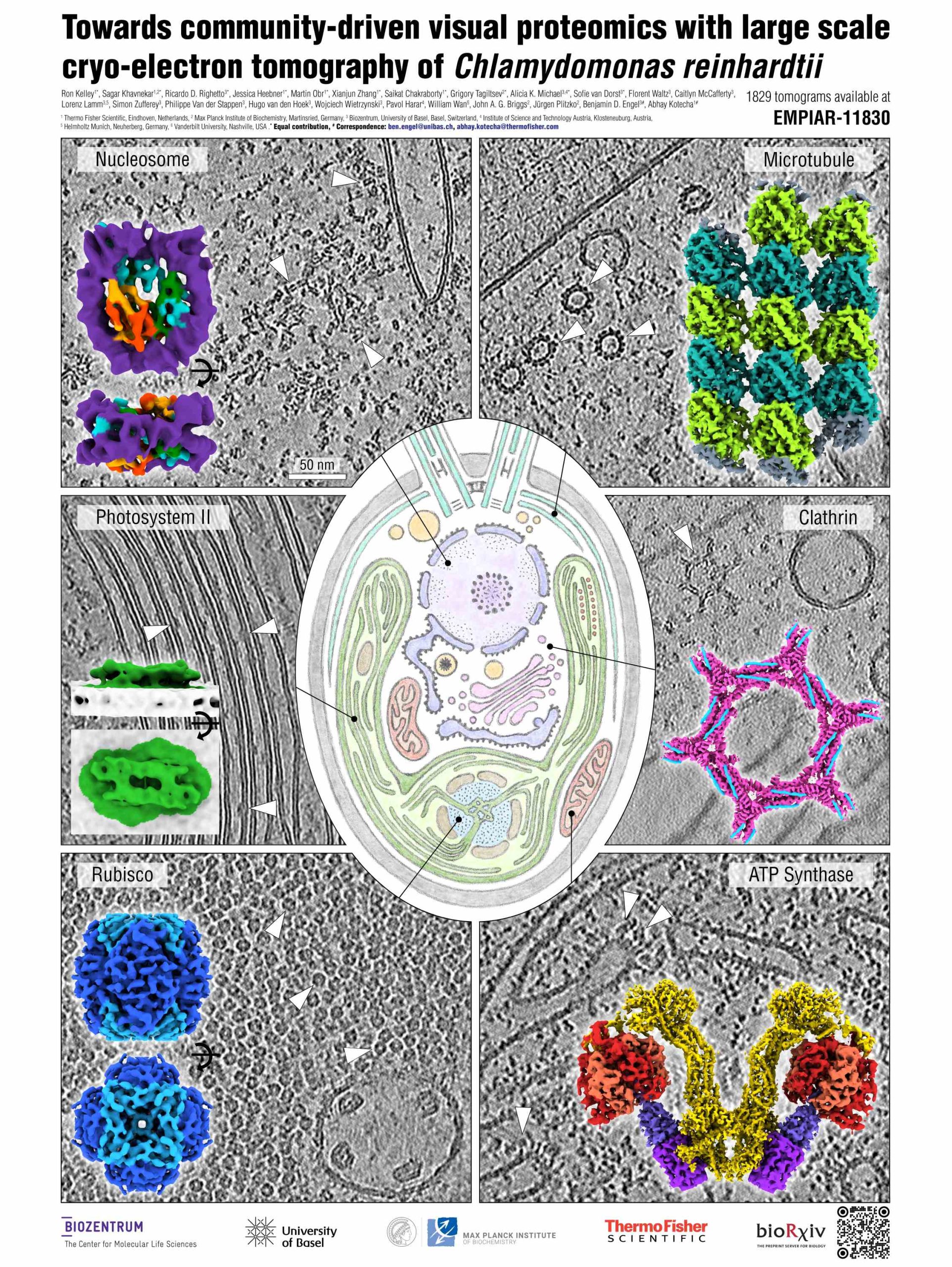
Towards community-driven visual proteomics with large scale cryo-electron tomography of Chlamydomonas reinhardtii
Presenter: Ricardo Righetto
Authors: Ricardo Righetto, Florent Waltz, Sagar Khavnekar, Caitlyn McCafferty, Ron Kelley, Lorenz Lamm, Martin Obr, Simon Zufferey, Jessica Heebner, Philippe Van der Stappen, Alicia K. Michael, Hugo Van Den Hoek, Xianjun Zhang, Wojciech Wietrzynski, Saikat Chakraborty, Paul Korir, Grigory Tagiltsev, Simone Weyand, Sofie Van Dorst, Aliaksei Chareshneu, John Briggs, Juergen Plitzko, Ben Engel, Abhay Kotecha

Abstract:
In situ cryo-electron tomography (cryo-ET) has emerged as the method of choice to investigate structures of biomolecules in their native context. Frozen cellular samples are thinned using cryo-focused ion beam milling (cryo-FIB) to produce ~150 nm sections, called lamellae. Conventional gallium-based cryo-FIB milling suffers from ice contamination, limited throughput, and manual transfers, which compromise both data quality and quantity. Here, we tested the application of a cryo-plasma-FIB instrument for high-throughput lamella milling of the green alga Chlamydomonas reinhardtii, a useful model system for in situ visualisation of numerous fundamental cellular processes. Combining cryo-plasma-FIB milling with recent advances in cryo-ET data acquisition and processing, we generated a dataset of 1829 reconstructed and annotated tomograms, which we provide as a community resource to drive method development and inspire biological discovery. To assay the quality of this dataset, we performed subtomogram averaging (STA) of both soluble and membrane-bound complexes ranging in size from >3 MDa to ~200 kDa, including the 80S ribosome, F1-ATP synthase, Rubisco, photosystem II, microtubules, and nucleosomes. The majority of these density maps reached sub-nm resolution, demonstrating the potential of this C. reinhardtii dataset, as well as the promise of modern cryo-ET workflows and open data sharing towards visual proteomics.
{kind=link}
Structurally heterogenous ribosomes cooperate in protein synthesis in bacterial cells
Presenter: Sophie Kopetschke
Authors: Karla Helena-Bueno, Sebastian Filbeck, Lewis Chan, Sonia Birsan2, Arnaud Baslé, Maisie Hudson, Stefan Pfeffer, Chris Hill, Sergey Melnikov, Sophie Kopetschke

Abstract:
Ribosome heterogeneity is a paradigm in biology, pertaining to the existence of structurally
distinct populations of ribosomes within a single organism or cell. This concept evokes the idea that structurally distinct pools of ribosomes have different functional properties and may be used to translate specific mRNAs. Whilst there is supporting evidence for conditional alterations in ribosomes structure, e.g. in response to environmental stimuli, it is unknown to what extent structural heterogeneity reflects genuine functional specialization rather than stochastic variations in ribosome assembly. To address this question, we combine high-resolution cryo-electron microscopy and tomography to directly observe structurally distinct ribosomes during protein synthesis in single bacterial cells. Specifically, we show that 70% of ribosomes in Psychrobacter urativorans contain a second copy of the ribosomal protein bS20 at a previously unknown binding site in the large ribosomal subunit. As a result, P. urativorans cells contain two structurally distinct types of ribosomes within a single cell. We then characterize individual and structurally distinct ribosomes to determine their active states, molecular partners, and their organization into polysomes in bacterial cells. This comprehensive analysis reveals that ribosomes with one or two copies of bS20 do not segregate into separate populations; instead, they function equivalently and cooperatively while co-occurring on the same mRNA molecules in situ. Overall, our work shows that, contrary to the prevailing hypothesis, ribosome heterogeneity does not necessarily lead to functional specialization, even when it involves significant variations, such as the presence or absence of a ribosomal protein. Instead, we show that the molecular structures of ribosomes possess previously unseen and functionally silent variations that allow
heterogeneous ribosomes to cooperate in general protein synthesis rather than translating discrete populations of mRNA.
Molecular architecture of synaptic vesicles
Presenter: Uljana Kravcenko
Authors: Uljana Kravcenko, Jana Kroll, Max Ruwolt, Artsemi Yushkevich, Martina Zenkner, Julia Ruta, Rowaa Lotfy, Erich Wanker, Christian Rosenmund, Fan Liu, Mikhail Kudryashev

Abstract:
Synaptic vesicles (SVs) play a crucial role in neurotransmission by storing and transporting neurotransmitters to the presynaptic active zone, where they are released via exocytosis. Following release, SV proteins and excess membrane can be recycled through endocytosis, enabling the formation of new SVs in a clathrin-dependent manner. This recycling process helps to maintain the complex molecular composition of SVs across multiple cycles. Previous research have investigated SV composition using mass spectrometry and fluorescence microscopy, resulting in a model of an average SV. However, the structural heterogeneity and molecular organization of individual SVs remain poorly understood. To address this, we used cryo-electron tomography to examine the molecular architecture of SVs isolated from mouse brains and within cultured neurons. Our findings revealed several classes of small proteins on the SV surface, as well as elongated proteinaceous densities inside the vesicles. We identified V-ATPases, determined their structure using subtomogram averaging, and demonstrated their colocalization with the small membrane-embedded protein synaptophysin (Syp). Using a bioluminescence assay, we detected pairwise interactions among vesicle-associated membrane protein 2 (VAMP2), Syp, and the V-ATPase Voe1 domain. Notably, V-ATPases were randomly distributed across the SV surface, independent of vesicle size. Additionally, a subset of SVs, both isolated and within neurons, exhibited partially assembled clathrin coats with a soccer ball symmetry. Some isolated clathrin-coated vesicles contained V-ATPases beneath the clathrin cages, suggesting that the V-ATPase V1 region may be recruited early in SV reformation. We also identified clathrin baskets lacking vesicles in both synaptic vesicle preparations and hippocampal neurons, and determined their location to be proximal to the cell membrane. Overall, our analysis provides new insights into the structural diversity and molecular organization of individual SVs.
The apoptosome assembly in situ
Presenter: Calvin Klein
Authors: Calvin Klein, Michael Wozny, Alicia Borgeaud, Thomas Lemmin, Wanda Kukulski

Abstract:
The apoptosome is a large protein complex crucial in initiating programmed cell death(apoptosis). It is a heptameric assembly of the apoptotic protease-activating factor 1 (Apaf1), and includes bound cytochrome c. The apoptosome forms upon initiating apoptosis, and recruits procaspase-9 to activate the caspase cascade, eventually leading to the dismantling of the cell. Although experimental structures of the apoptosome are available, the occurrence of the complex has yet to be shown in situ. Using Cryo-Correlative Light Electron Microscopy (Cryo-CLEM) our lab has found that dense meshworks in the cytosol of apoptotic cells correlate with foci of Apaf1-GFP. Cell biological data indicate that these foci are functional apoptosome equivalents [1]. Based on these findings, we aim to uncover the underlying structure of these Apaf1 dense meshworks within cells. To further investigate the Apaf1 dense meshwork, we currently increase our dataset of electron cryo-tomograms of cryo-FIB milled cells. Additionally, we use a modified protocol to unroof cells on cryo-ET grids, obtaining thin samples of the dense meshwork in a close-to-native environment, without the need for previous FIB-milling [2]. At the same time, we are testing a computational pipeline to analyze the amorphous meshwork structurally. Our goal is to detect substructures within the meshwork using tools such as Rasterized Subtomogram Extraction (RSE), subtomogram classification, and subtomogram averaging (STA). At present, we acquired a dataset of 12 tomograms of cryo-FIB milled cells and 52 tomograms of unroofed cells showing the Apaf1 dense meshwork. We used RSE to extract subtomograms within the regions of interest and are classifying potential subunits of the Apaf1 dense meshwork by applying the latest Relion 5 subtomogram averaging pipeline.
Due to the confidentiality of the unpublished data, we cannot share the poster.
An improved cryo-FIB-ET workflow towards quantitative cryo-electron tomography
Presenter: Sebastian Tacke
Authors: Elisa Lisicki, Tatjana Taubitz, Stefan Raunser, Mingjun Xu, Sebastian Tacke

Abstract:
In recent years, cryo-electron tomography (cryo-ET) has become a versatile tool to qualitatively describe the ultrastructure of cellular systems and characterize previously elusive protein complexes in their native context. The beauty of cryo-ET is the wealth of information which can be visualized within a single tomogram. In theory, cryo-ET data can be utilized not only to visualize and analyze the protein complex in-situ, but also to study protein-protein and protein-organelle interaction, the full proteome and its interaction. To unveil the full complexity captured in a dataset, a quantitative analysis is essential. This requires a sufficiently large dataset, and each step of the complex cryo-ET workflow to be as efficient as possible. Despite notable improvements in sample preparation, post-processing, sample handling, and data acquisition in recent years, several challenges remain. First, the complex cryo-ET workflow often demands the samples to be stored either for further processing or for final imaging. Although improvements were made, ice contamination during handling and storage still limits the overall efficiency of the preparation pipeline. Secondly, current preparation protocols do not make use of high currents, limiting the throughput, especially for thick samples like organoids or tissues. Here we present solutions to overcome the aforementioned limitations. To avoid sample contamination, we have developed an enhanced glove box system that allows sample handling and storage in a contamination free environment. With these technical improvements, we can now almost completely prevent ice contamination of the polished lamellae during transfer, handling and storage, enabling a flexible and efficient cryo-ET pipeline. Secondly, we benchmarked the impact of high currents ( > 60nA) on the sample. Our results clearly show that even currents as high as 2.5μA can be used for sample preparation if used correctly. After characterizing the effects of milling with these high currents, we were able to significantly reduce the milling time for thick samples like organoids and tissues. Taken together, we have enhanced the overall efficiency of the cryo-ET pipeline, allowing the production of sufficiently large datasets for detailed quantitative analysis.

Find out more about the #EMBOinsitu workshop from the blog post written by Ahmed Adel Ezat, who participated as an event reporter!
The EMBO Workshop ‘In situ structural biology: expanding the toolbox for structural cell biology‘ took place from 4 – 7 February 2025 at EMBL Heidelberg and virtually.