Bioinformatics tutorial
- Overview
- Sequence preparation – part 1
- Sequence preparation – part 2
- Sequence preparation – part 3
- Database search tutorial
- Activity navigation
Overview
This page provide a tutorial on using your DNA sequencing data to identify your plant species using various bioinformatics tools.
If you would like to work alongside the tutorial with a real-life barcoding sequence, you can download the example sequence data here.
Sequence preparation – part 1
This tutorial will guide you through the process of preparing a sequence for the search against database entries contained in the European Nucleotide Archive (ENA).
Imagine we have extracted DNA from an unknown plant species (called “TutorialExample”), amplified the rbcL barcode and submitted the PCR products for sequencing to an external service.
Sequence information
Imagine we have received a forward and reverse sequence of the plant DNA barcode from the sequencing service. This information is provided in three different types of files: .ab1, .seq, and .fas. The first two files will be used during this project. While .ab1 files store visual information about the sequence (more details later on), .seq files store sequence information in a text format called FASTA.
In FASTA format, each sequence starts with a line containing a greater-than sign (>) and a descriptive header (this is the name of the sequence). The name of the sequence is added without introducing a space. The following image shows the forward and reverse sequence of the “TutorialExample” in FASTA format. The “>” sign is highlighted in pink, the descriptive header is shown in blue and the sequence is depicted in brown.

Assembling a consensus sequence
Before the identity of the unknown plant species can be established by searching the barcode against the entries in ENA, the sequences of the forward and reverse reads have to be assembled into a single consensus sequence called a contig.
Assembling the contig of the DNA barcode will involve the following steps:
1. converting the reverse sequence into its reverse complement
2. aligning the forward and reverse sequence reads
3. editing and assembling the consensus sequence
Go through the instructions in the tabs below (left to right) and learn how to assemble a contig.
Reverse Complement
To be able to align the forward and reverse sequence reads, the two sequences have to have the same orientation. This can be achieved by converting the reverse sequence into its reverse complement (i.e. converting the 3′-5′ sequence into a 5′-3′ orientation).
Forward and reverse reads – definitions in the context of sequencing:

Unlike PCR amplification – which requires a forward and a reverse primer – Sanger sequencing only needs a single primer. However, as a single primer might not produce reliable cover for the whole sequence (particularly towards the 3′ end), sequencing is usually carried out with at least two primers. In this project, we use a universal primer pair (M13 sequencing primers) to sequence the barcode separately from both ends, thus the M13 primers are termed forward and reverse primers. In two separate, independent reactions, the forward primer synthesises a strand 5′ to 3′ from the 3′ end of one strand (the forward read), and the reverse primer synthesises a strand 5′ to 3′ from the 3′ end of the complementary strand (the reverse read).
Complement and reverse complement – definitions:

To be able to align the forward and reverse sequence reads, the two sequences have to have the same orientation. This can be achieved by converting the reverse sequence read into its reverse complement (i.e. converting the 3′-5′ sequence into a 5′-3′ orientation). The example below illustrates the principle of reverse-complementing a reverse read.
The following steps are carried out in order to convert the reverse sequence read into its reverse complement:
1. Open the .seq file of your reverse sequence using a text editor such as NotePad on Windows or TextEdit on Mac. The .seq file contains the sequence information in FASTA format.
2. Copy the whole sequence including the “>” sign and descriptive header (keyboard shortcut Ctrl + C).
3. Paste all the information into the EMBOSS Seqret input box below (Ctrl + V). Alternatively, use the upload function to upload the reverse .seq file.
4. In “Step 1” ensure “DNA” is selected as input data. In “Step 2” select “FASTA format” as input and output format. To receive the reverse complement of the sequence, click on “More options” and select “Yes” as “Reverse” option.
To view the EMBOSS Seqret search window containing “TutorialExample” sequence and correct parameter settings click here.
5. Click on “Submit”.
6. Open an empty text editor document on your computer.
7. Once your reverse complement sequence is available in the “Tool Output” window of EMBOSS Seqret, copy the whole sequence into the new text editor document (Ctrl + C and Ctrl + V). Again, include the “>” sign and descriptive header when copying the sequence. To view the EMBOSS Seqret results window showing the “TutorialExample” sequence in reverse complement click here.
8. Keep the “>” sign at the beginning sequence information but replace the descriptive header by “SampleID_RP_RevComp”. Save the new text editor document as “SampleID_RP_RevComp” on your desktop.
If you would like to use an already prepared reverse complement sequence of the “TutorialExample” (rather than the one you just prepared), you may download it here.
Proceed to the next tab to learn how to align the forward and reverse sequence.
Sequence preparation – part 2
Alignment
The reverse sequence which was reversed and complemented in the last step can now be aligned with the forward sequence. The alignment will reveal the consensus sequence as well as any nucleotide mismatches or gaps between the forward and reverse reads. Nucleotide positions with mismatches or gaps can then be cross-checked with the chromatogram-view of the forward and reverse sequences and a single consensus barcode sequence can be assembled.
The following steps are carried out in order to align the forward and reverse sequence reads and assemble a contig:
1. Open the .seq file of your forward sequence. Copy and paste the whole sequence including the “>” sign and descriptive header (keyboard shortcut Ctrl + C) into the first EMBOSS Needle input box below.
2. Open the text file “SampleID_RP_RevComp”. Copy the whole sequence including the “>” sign and descriptive header (keyboard shortcut Ctrl + C and Ctrl + V) into the second EMBOSS Needle input box below.
Alternatively, use the upload function to upload the forward sequence and the edited reverse sequence.
3. Keep the default settings in “Step 2” and click on “Submit”. To view the EMBOSS Needle search window containing “TutorialExample” sequence and correct parameter settings click here.
4. Once the alignment is available, click on “View alignment file” and scroll down to study the sequence alignment. The alignment of the “TutorialExample” is shown below. To view the full EMBOSS Needle alignment result window click here.

Guide to EMBOSS Needle nucleotide sequence alignment result:
Vertical lines indicate identical nucleotides (matches).
Dots indicate mismatches (i.e. conflicting nucleotide reads in forward and reverse sequence).
Empty spaces indicate gaps (i.e. a nucleotide read versus no nucleotide read in the two sequences) or mismatches.
Horizontal dashes within a sequence indicate gaps.
Horizontal dashes at either side of the alignment indicate that the two sequences did not produce an overlapping read in these areas (no alignment).
The letter “N” within a sequence is used to denote an unspecified nucleotide.
5. Open the chromatograms of your forward and reverse sequence by opening the respective .ab1 files using a chromatogram viewer (see “Chromatogram” tab). For easier analysis, reverse complement the reverse chromatogram (Chromas Lite: “Edit” > “Reverse+Complement”; 4Peaks: “Edit” > “Flip sequence”).
A sequencing chromatogram displays the data produced by the sequencing machine as a so-called trace. Analysing the chromatograms of your forward and reverse sequences will help you to check the quality of the sequences and to cross-check mismatches or gaps identified via the forward-reverse sequence alignment. Guidelines on how to interpret chromatogram information can be found at the “Chromatogram” tab.
To view excerpts of the forward and reverse chromatograms of the “TutorialExample”, click here:
- Forward read: first 100 base pairs; final 100 base pairs
- Reverse read, reverse complemented: first 100 base pairs; final 100 base pairs
6. Now prepare a document which will hold your contig sequence in FASTA format. Open an empty text editor document on your computer. Copy the whole forward sequence from your .seq file into the new text document (Ctrl + C and Ctrl + V). Keep the “>” sign at the beginning sequence information but replace the descriptive header by “SampleID_Contig”. Save the document as “SampleID_Contig” on your desktop.
7. Go through the alignment and identify gaps or mismatches. For every mismatch or gap, go to the respective nucleotide position in the forward and reverse chromatograms (you can use the search function of the software to find the position within the sequence). [Note that the nucleotide numbering of the alignment does not necessarily correspond to the numbering of the chromatograms as the FASTA might have been truncated by the sequencing service already.] Looking at the two chromatograms, compare the peaks at the respective nucleotide position and decide whether the forward or reverse read looks more reliable. You might also be able to identify the identity of any “unknown” nucleotides (“N”).
In your “SampleID_Contig” text document edit the sequence according to your analysis (remember that you have copied the forward sequence).
For the purpose of contig assembly, the EMBOSS Needle alignment together with the chromatogram of the “TutorialExample” were interpreted as follows (cf. images of chromatograms and FASTA sequence below):
A total number of 6 gaps and mismatches were identified (4 gaps, 2 mismatches).
Mismatch/gap 1:
- FASTA file information: forward: N (unknown), reverse: T
- Forward chromatogram: (a) nucleotide position at beginning of sequence within area of irregular peaks; (b) T appears as part of a double peak (i.e. that the sequence analysis software did not feel confident enough to transfer the signal into FASTA format).
- Reverse chromatogram: (a) nucleotide position in middle part of sequence within area of regular, well-spaced peaks without background; (b) T gives sharp, well-separated peak.
- Decision: the unknown nucleotide appearing in the forward read can be identified as T.
Mismatch/gap 2:
- FASTA file information: forward: – (gap), reverse: A
- Forward chromatogram: (a) nucleotide position at beginning of sequence within area of irregular peaks; (b) two A’s appear as part of a double peak.
- Reverse chromatogram: (a) nucleotide position in middle part of sequence within area of regular, well-spaced peaks without background; (b) an additional A appears as sharp, well-separated peak.
- Decision: an additional A is added in front of the two existing A’s of the forward sequence.

Mismatch/gap 3:
- FASTA file information: forward: – (gap), reverse: A
- Forward chromatogram: (a) nucleotide position towards the end of the sequence within area of well-separated but fairly low peaks; (b) there are 5 well-separated peaks of A’s.
- Reverse chromatogram: (a) nucleotide position towards the beginning of the sequence within area of well resolved peaks; (b) there are 5 well-separated peaks of A’s.
- Decision: as both chromatograms show the traces of 5 rather than 6 A’s, we can be confident to keep a sequence of 5 A’s.

Mismatch/gap 4:
- FASTA file information: forward: – (gap), reverse: N
- Forward chromatogram: (a) nucleotide position towards the end of the sequence within area of peaks which are close but not overlapping and fairly low; (b) no peaks of additional nucleotides visible.
- Reverse chromatogram: (a) nucleotide position at the beginning of the sequence within area of irregular and overlapping peaks; (b) overlapping peaks at the position of “N”.
- Decision: the reverse read at this position is not reliable; no additional nucleotide is added.
Mismatch/gap 5:
- FASTA file information: forward: G, reverse: N
- Forward chromatogram: (a) nucleotide position towards the end of the sequence within area of peaks which are close but not overlapping and fairly low; (b) peak for G clearly visible without background.
- Reverse chromatogram: (a) nucleotide position at the beginning of the sequence within area of irregular and overlapping peaks; (b) peaks overlapping, resulting in sequence analysis software unable to confidently identify nucleotide.
- Decision: the unknown nucleotide appearing in the reverse read can be identified as G.
Mismatch/gap 6:
- FASTA file information: forward: T, reverse: C
- Forward chromatogram: (a) nucleotide position towards the end of the sequence within area of peaks which are close but not overlapping and fairly low; (b) peak for T clearly visible without background.
- Reverse chromatogram: (a) nucleotide position at the beginning of the sequence within area of irregular and overlapping peaks; (b) overlapping peaks at the position of C.
- Decision: the nucleotide is identified as T.


8. Once you have completed all the necessary edits, you have assembled the contig of your sample’s barcode. Make sure you save the document!
If you would like to use an already prepared contig of the “TutorialExample” (rather than the one you just prepared), you may download it here.
The plant barcoding sequence is now ready to be searched against the entries in ENA.
Sequence preparation – part 3
Chromatogram
A sequencing chromatogram displays the data produced by the sequencing machine as a so-called trace. Each of the four nucleotides is represented by a different colour (A = green, C = blue, G = black, T = red) and the sequence can be “read” as a sequence of individual peaks.
Chromatograms are stored as .abi or .ab1 files and can be viewed using specialist software. The chromatogram viewers we recommend for this project are listed here (“chromatogram” tab).
Analysing the chromatograms of your forward and reverse sequence reads will help you to check the quality of the sequences and allows you to cross-check mismatches or gaps identified via the forward-reverse sequence alignment.
Basic guidelines on how to recognise a good quality sequence
The first 30 or so peaks on either side of the sequence are usually not very good (e.g. broad, round (“rolling hills”), crowded peaks, peak overlaps or peaks not clearly resolved from each other). This is a normal occurrence and the sequencing service used during this project already trims the FASTA-version of the sequence to remove these unreliable reads, so it will not affect the quality of your data.
In the middle of the chromatogram a good quality sequence should have:
- peaks which are higher than at the ends of the sequence
- peaks which are sharp and well resolved/separated from each other, with even spacing between peaks (i.e. you should be able to clearly read the sequence off the peaks manually).
- little or no background peaks at the baseline

Database search tutorial
This tutorial will guide you through the process of identifying nucleotide sequences in the European Nucleotide Archive (ENA) which match your barcode sequence. An rbcL barcode sequence of an unknown plant species (called “TutorialExample”) is used as an example.
In order to submit an ENA search, the forward and reverse sequencing reads of the sample have to be assembled into a contig. Visit the “Sequence preparation tutorial” tab for information on how this is done.
The following steps describe how to search ENA and how to interpret the search result:
1. Copy your whole contig sequence from the text file “SampleID_Contig”, including the “>” sign and descriptive header, into the ENA search box below (keyboard shortcut Ctrl + C and Ctrl + V). Alternatively, use the upload function to upload your text file.
2. In the field “Search against” select ” Assembled and annotated sequences” and “Limit sequence by” > “Data class” > “Standard sequences (STD)”.
3. Initiate the search by clicking on “Submit” (you might need to scroll back to the left to see the “Submit” button). The inserted sequence will now be compared to all the known sequences contained in the database and the best alignment hits will be displayed.
4. In the “Summary table” you will see the top 50 sequence search results.
By default, the search results are sorted according to their “Score”, with the highest at the top. Results may also be sorted according to any other value of the results columns by clicking on the up/down arrows. However, for the purpose of identifying the closest match, keep the results sorted according to “Score”.
5. To identify the best match, proceed as follows: sort the search results according to their score (highest at the top), if not done already. The result with the combined highest score and lowest E-value is your best match. In case there are multiple results which have the combined highest score and lowest E-value, choose the one with the highest identity percentage.
In case there are two or more results with identical score/E-value/% identity, the database is unable to discriminate between the entries (e.g. due to inaccuracies in your input sequence) or might not contain an entry of your species. If this is the case, record all of your top results. You might still be able to identify your sample to genus level.
With respect to the “TutorialExample”, the top 10 entries all display identical E-values, scores, % identity and % positives. These values do not allow to discriminate between the results, making it impossible to identify one of the 8 species listed as matching with the “TutorialExample”. This means that identification is only possible on genus (rather than species) level.
The image below shows the ENA search result of a second, unrelated, example. The species of this sample can be readily identified by the database: Although the E-values of the top 10 results is identical, the results can be discriminated by looking at the score (as well as % identity). With a highest score and highest % identity, alignment number 1 is the closest match to the sample barcode.
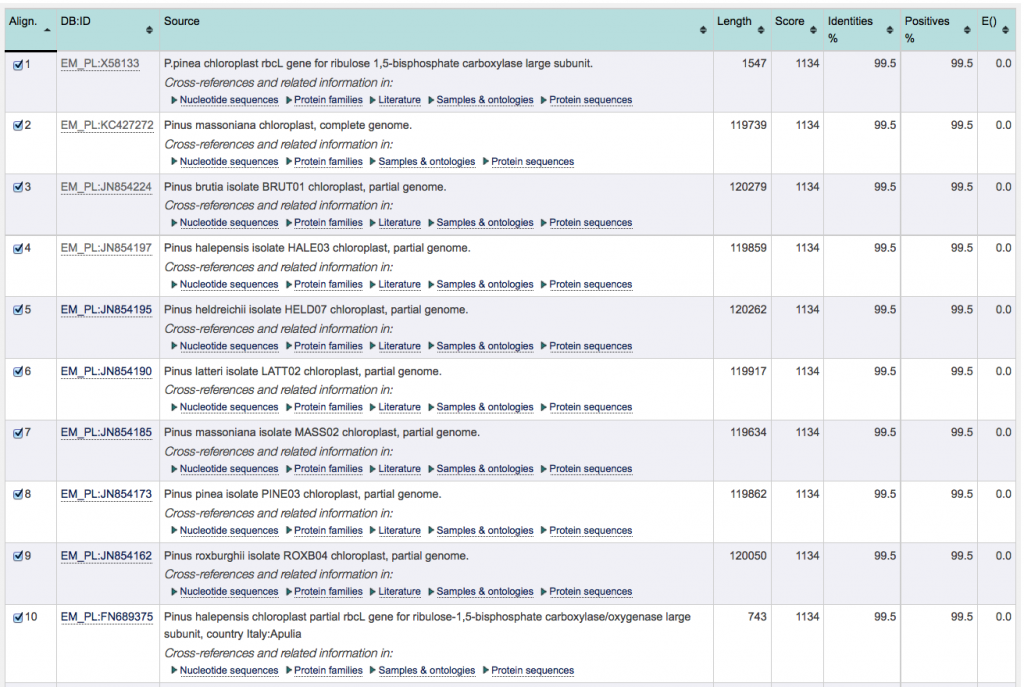
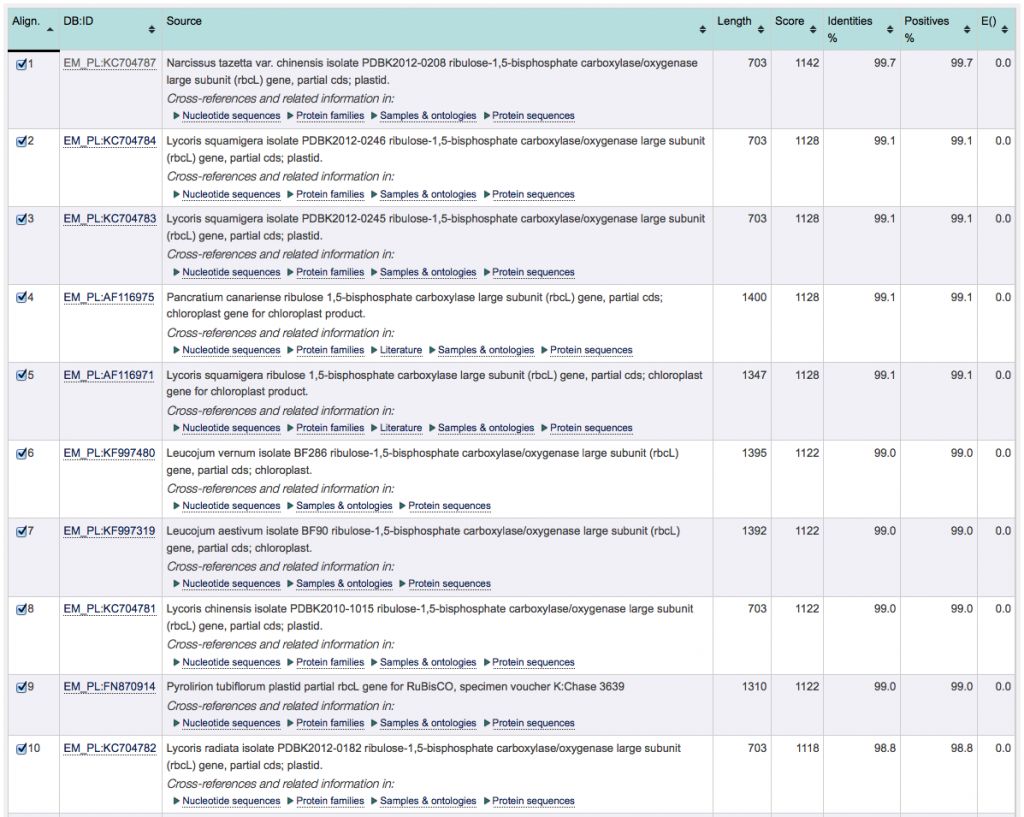
6. Can you identify the best database match for your sequence? Which organism does it belong to? Can you identify your sample to genus, or even species, level?
You have now identified the genus and, possibly, species name of your plant sample.
Activity navigation
Sample collection and wet-lab
Bioinformatics

Share: