English
English Čeština
Čeština Ελληνικά
Ελληνικά
TreeTOPS – Ein Phylogenetik-Icebreaker Spiel
Einführung
Das übergeordnete Ziel des Spieles ist es, die Spieler in das Thema Phylogenetik, also in die Lehre von evolutionären Verwandtschaftsverhältnissen zwischen taxonomischen Gruppen (z.B. Organismen, Arten und Populationen) oder anderen biologischen Einheiten (z.B. Genen und Proteinen), einzuführen. Die Spieler spielen in Gruppen und bauen mit Hilfe kleiner Farbschemata-Spielkarten einen phylogenetischen Baum, der den evolutionären Verzweigungsprozess einer imaginären taxonomischen Gruppe oder biologischen Einheit darstellt.
Hintergrund und Spielanleitung
Klassische und Molekulare Phulogetetik
Die phylogenetische Rekonstruktion spielt eine Schlüsselrolle für das Verständnis von evolutionären Prozessen. Traditionelle Klassifizierungsmethoden beschäftigen sich mit den evolutionären Beziehungen von klassischen biologischen Einheiten wie Stämmen, Arten oder Populationen und stützen sich dabei auf morphologische Beobachtungen und phänotypische Charakteristiken. Die rasanten Entwicklungen in der Molekularbiologie innerhalb der vergangenen Jahrzehnte hat phylogenetische Untersuchungen von Organismen sowie von Molekülen jedoch revolutioniert. Techniken wie die Polymerase-Kettenreaktion (PCR), Restriktions-Fragment-Längen-Polymorphismus (RFLP) und das Sequenzieren von ganzen Genomen, gekoppelt mit einer Zunahme an biologischen Datenbanken, hat den Anwendungsbereich der Phylogenetik bedeutsam erweitert. Dies hat es Wissenschaftlern ermöglicht evolutionäre Stammbäume zu erstellen, die auf dem molekularen Aufbau der Organismen beruhen. Durch diese Entwicklungen sind molekulare Daten mittlerweile zur Hauptquelle für die Konstruktion von Stammbäumen geworden.
Es gibt zahlreiche Beispiele dafür, wie Stammbäume, die auf der Basis molekularer Daten erstellt wurden, Evolutionsforschung vorangetrieben haben. Zum Beispiel wurden Pilze bis in die 1970er Jahre als ein Unterreich des Pflanzenreichs eingeordnet. Die Analyse molekularer Daten zeigte jedoch, dass Pilze tatsächlich näher mit Tieren als mit Pflanzen verwandt sind. Diese Erkenntnis führte zu einer drastischen Umklassifizierung der Pilze in ihr eigenes Reich. Molekulare Sequenzdaten haben es Wissenschaftler des weiteren ermöglicht, ein Klassifizierungssystem für z.B. Viren, Gene und Proteine zu erstellen – Erkenntnisse die wichtige Einblicke in verschiedenste Forschungsfelder – von Evolutionsbiologie über Proteomik zu Medizin – erlauben. Durch das Verwenden molekularer Daten für die Bewertung von Verwandtschaftsverhältnissen ist die Bestimmung taxonomisch gemeinsamer Vorfahren präziser geworden. Diese Entwicklung gestattet nicht nur grundlegende Hinweise auf evolutionäre Verwandtschaftsverhältnisse von Organismen, sondern hilft auch bei der Bestimmung von neuen Modellorganismen oder von Organismen, die einem ausgestorbenen gemeinsamen Vorfahren ähnlich sind.
Stammbaum-grundlagen
In phylogenetischen Bäumen wird der Grad evolutionärer Verwandtschaft durch die Positionen der einzelnen Einheiten (z.B. Arten, Populationen, Gene) zueinander bestimmt. Einheiten, welche evolutionsgeschichtlich nah miteinander verwandt sind werden im Stammbaum nah zueinander platziert; Einheiten, welche entfernter verwandt sind werden mit größeren Abständen auf anderen Zweigen oder Kladen platziert. Im folgenden Text werden zwei der häufigsten phylogenetischen Bäume verglichen und einige wichtige Begriffe erklärt.
Ungewurzelte und gewurzelte phylogenetische Bäume
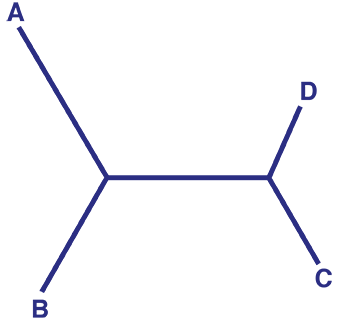
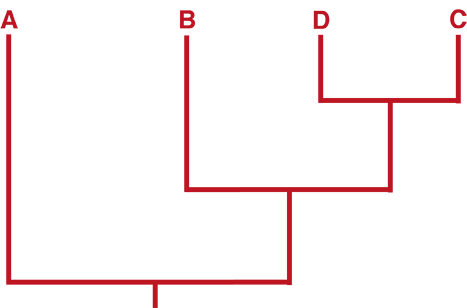
Ein ungewurzelter Baum bietet keine Informationen über den gemeinsamen Vorfahren der untersuchten Einheiten. Ein ungewurzelter Baum kann in einen gewurzelten Baum umgewandelt werden – dieser bietet Informationen über den gemeinsamen Vorfahren (Wurzel).
Vergleicht man das untersuchte Merkmal der untersuchten Gruppe (ingroup) mit einem Merkmal einer weiter entfernt verwandten Gruppe (outgroup), so kann das Ergebnis darüber Aufschluss geben, ob das untersuchte Merkmal ursprünglich oder abgeleitet ist.
Phylogenetische Begriffsdefinitionen für gewurzelte Bäume
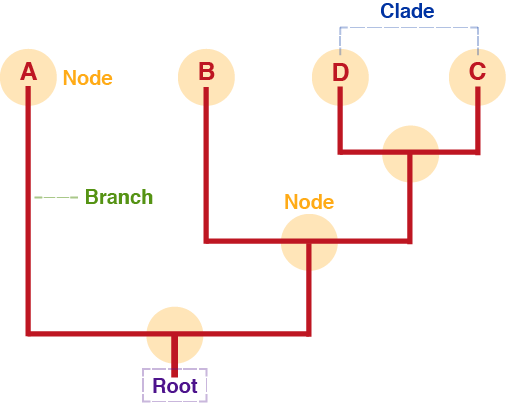
Zweig
Er verbindet die Knoten des Baumes und repräsentiert den vorgeschlagenen Evolutionsverlauf der untersuchten Linie.
Wurzel
Ein Knoten, der den gemeinsamen Vorfahren aller Knoten des Baumes repräsentiert.
Knoten
Innerer Knoten: repräsentiert einen hypothetischen Vorfahren des Taxons (d.h. Population, Art, Gen, Protein, usw.).
Äußerer Knoten/Endknoten (Blatt): repräsentiert ein (normalerweise heutige auftretendes) Taxon (d.h. Population, Art, Gen, Protein, usw.), das mit anderen Taxa im Baum verglichen wird.
Klade
In einer Klade haben alle Mitglieder einen gemeinsamen Vorfahren. Dieser Vorfahre ist kein gemeinsamer Vorfahre anderen Baum-Mitglieder, die sich außerhalb der Klade befinden.
Herausforderungen bei der stammbaum-erstelung
Wenn man beurteilen möchte, wie bestimmte biologische Einheiten miteinander verwandt sind, stellen die hypothetischen Evolutionsprozesse wie parallele, konvergente und vernetzte (retikulate) Evolution meist die größten Herausforderungen dar. Beim Erstellen des Stammbaumes muss daher in Betracht gezogen werden, dass solche Prozesse eventuell auf die untersuchte Gruppe eingewirkt haben könnten. Der folgende Text bietet kurze Definitionen dieser drei Evolutionsprozesse.
Parallele Evolution
Parallele Evolution beschreibt einen Prozess in dem Organismen, Taxa, oder andere biologische Einheiten (z.B. Gene oder Proteine), die von einem gemeinsamen Vorfahren abstammen, unabhängig voneinander vergleichbare (analoge) Merkmale entwickelt haben. Parallele Evolution kann z.B. das Ergebnis von Anpassung (Adaption) an z.B. die gleiche ökologische Nische sein, oder durch andere selektive Drucks zustande kommen.
Konvergente Evolution
Konvergente Evolution beschreibt einen Prozess in dem Organismen, Taxa, oder andere biologische Einheiten (z.B. Gene oder Proteine), die keinen gemeinsamen Vorfahren haben, unabhängig voneinander vergleichbare (analoge) Merkmale entwickelt haben. Konvergente Evolution kann z.B. das Ergebnis von Anpassung (Adaption) an z.B. die gleiche ökologische Nische sein, oder durch andere selektive Drucks zustande kommen.
Vernetzte (retikulate) Evolution
Vernetzte Evolution beschreibt einen Prozess in dem sich genetisch unterschiedliche Linien/Arten rekombinieren und eine neue Art entsteht, die reproduktiv isoliert von der Ausgangsart ist. Dieser Prozess der Artbildung wird Hybrid-Artbildung genannt. Falls eine strikt hierarchische Struktur eines Stammbaumes die evolutionären Beziehungen der untersuchten Gruppe nicht angemessen widerspiegeln würde, sollte bei der Stammbaumerstellung in Erwägung gezogen werden, dass eventuell Prozesse der vernetzte Evolution auf die untersuchte Gruppe eingewirkt haben.
Wie bei der Analyse von Verwandtschaftsverhältnissen klassischer biologischer Einheiten wie Arten oder Populationen, werde ähnliche Methoden auch in Studien zur Evolution von Genen und Proteinen angewandt. Hierzu werden homologe DNA- bzw. Aminosäure-Sequenzen und -Funktionen miteinander verglichen. Diese Methode kann interessante Einblicke in die evolutionären Beziehungen von Genen bzw. Proteinen liefern. Homologe Sequenzen haben identische oder sehr ähnliche Sequenzen und einen gemeinsamen Ursprung. Die Tatsache, dass es mehrere Prozesse gibt durch die Homologe entstehen können, erschwert das Bewerten von Verwandtschaftsverhältnissen. Das heißt, es ist auch in diesem Fall wichtig, die verschiedenen evolutionären Prozesse, die auf die untersuchte Gruppe eingewirkt haben könnten, nicht außer Acht zu lassen. Im Folgenden werden die zwei häufi gsten Homolog-Arten beschrieben.
Ortholog
Homologe biologische Einheiten (z.B. Gene oder Proteine) werden als ortholog bezeichnet wenn sie in verschiedenen Arten vorkommen und von der gleichen biologischen Ur-Einheit (gemeinsamer Vorfahre) abstammen. Orthologe entstehen durch Artbildung eines gemeinsamen Vorfahrens und haben normalerweise im Laufe der Evolution die gleichen oder ähnliche Merkmale behalten.
Paralog
Homologe biologische Einheiten (z.B. Gene oder Proteine) werden als paralog bezeichnet wenn sie in der gleichen Art (oft im gleichen Organismus) vorkommen und durch (kürzliche oder historische) Gen-Duplikation innerhalb dieser Art/Organismus entstanden sind. Normalerweise unterscheiden sich Paraloge im Laufe der Zeit in ihrer Funktion (und manchmal auch Sequenz).
Spielmaterial
Downloaden Sie das Spielmaterial als pdf-Datei here:
- Lehrerhandbuch mit Informationen zu Hintergrund, Spielanleitung und Lösungen
- Spielkarten
Spielanleitung
Spielmaterial
- Lehrerhandbuch mit Informationen zu Hintergrund, Spielanleitung und Lösungen
- Spielkarten (auf druckfähigen Magnetschildern oder laminiertem Papier)
- Magnetisches Whiteboard/Tafel bzw. Papier-bezogene Pinnwand
- Whiteboard-Stift/Kreide bzw. Stift für Papier
Spieler
Jedes der drei TreeTOPS Spiele kann von einer variablen Spielerzahl gespielt werden – von einer einzigen Person bis zu einer größeren Gruppe. Falls die Spiele zu Team-Building Zwecken genutzt werden sollen, kann TreeTOPS, je nach Spiel (Spiel 1-3), mit einer Gruppe von 8-9 bzw. 7-8 Spielern gespielt werden.
Ziel des Spieles
Jede Spielkarte stellt eine imaginäre biologische Einheit dar. Das Ziel des Spieles ist es, die Karten nach Verwandtschaftsgrad zu ordnen und mit den “Nachfahre“-Karten einen gewurzelten phylogenetischen Stammbaum zu erstellen, wobei die “Gemeinsamer Vorfahre“-Karte die Wurzel bildet. Dabei sollte beachtet werden, dass es verschiede Möglichkeiten gibt, den Stammbaum zu bauen; es also nicht eine einzige richtige Lösung gibt. Das Ziel des Spieles ist erreicht, wenn die Spieler ihre Entscheidungsgründe zu den jeweiligen Positionen der “Nachfahren” im Baum plausibel darlegen können.
Hinweis
Wenn es darum geht, die “Hierarchien” und “Gruppierungen” innerhalb des Baumes festzulegen ist es hilfreich, sich daran zu erinnern, dass Evolution auf der genetischen Ebene durch kleine Veränderungen wie Deletionen (Entfernungen), Dublikationen (Vermehrungen), Insertionen (Einschübe) und Substitutionen (Austausch) von Nukleotiden oder Nukleotid-Abschnitten geschieht.
Spielaufbau
Alle Spielkarten (“Nachfahre“-Karten und “Gemeinsamer Vorfahre“-Karte) werden an die Spielgruppe verteilt.
Falls TreeTOPS für Team-Building Zwecke genutzt werden soll, kann der Aufbau so aussehen:
8 (7) Spieler: Jeder Spieler erhält eine “Nachfahre“-Karte. Die “Gemeinsamer Vorfahre“-Karte wird am unteren Ende des Spielbrettes platziert und dient als Ausgangspunkt des Stammbaumes.
8 (9) Spieler: Ein Spieler erhält die “Gemeinsamer Vorfahre“-Karte, die restlichen Spieler erhalten jeweils eine “Nachfahre“-Karte.
Spielverlauf
Falls TreeTOPS für generelle Zwecke genutzt wird, verläuft das Spiel nach den Schritten 1a, 2-4. Falls TreeTOPS spezifisch für Team-Building Zwecke genutzt wird, verläuft das Spiel nach den Schritten 1b, 2-4.
1a) Die “Gemeinsamer Vorfahre”-Karte wird auf dem Brett platziert. Die Spieler gruppieren die “Nachfahre“-Karten nach deren Verwandtschaftsverhältnissen.
1b) Der Spieler mit der “Gemeinsamer Vorfahre”-Karte platziert seine Karte auf dem Brett. Die Spieler mit den “Nachfahre“-Karten gruppieren sich im Raum nach den Verwandtschaftsverhältnissen ihrer Spielkarten. Der Spieler mit der “Gemeinsamer Vorfahre”-Karte darf den anderen Spielern bei deren Gruppierung behilfl ich sein und kann ggf. als Koordinator wirken.
2) Die Spieler platzieren ihre “Nachfahre“-Karten in den festgelegten und mit allen abgestimmten Positionen auf dem Brett.
3) Die Spieler zeichnen einen phylogenetischen Stammbaum in dem sie die einzelnen Spielkarten verbinden.
4) Sobald der Stammbaum fertiggestellt ist, erklären die Spieler den anderen Spielergruppen bzw. ihrem Lehrer was sie dazu bewegt hat, den Stammbaum in dieser Art zu bauen.
Galerie




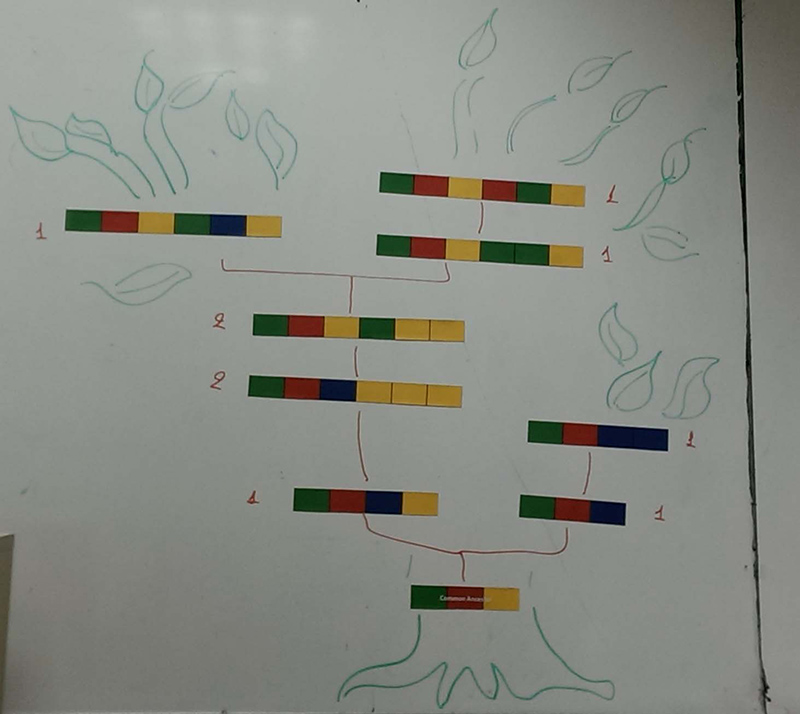
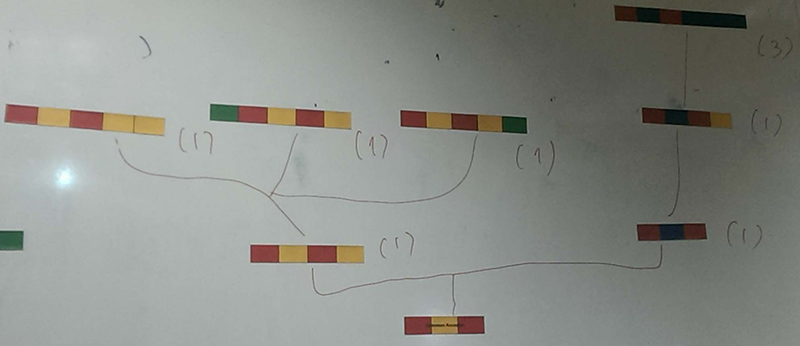

Topic area: Evolutionary biology
Age group: 16-19
Author: ELLS Team
Share: