Our research highlights the pivotal role of structural variants (SVs)—such as deletions, insertions, and inversions—which account for the majority of polymorphic bases in human genomes. Recent advancements in our lab show how somatic SVs accumulate throughout human life, which challenges the traditional notion of genomic stability within individuals, with significant implications for aging and disease susceptibility.
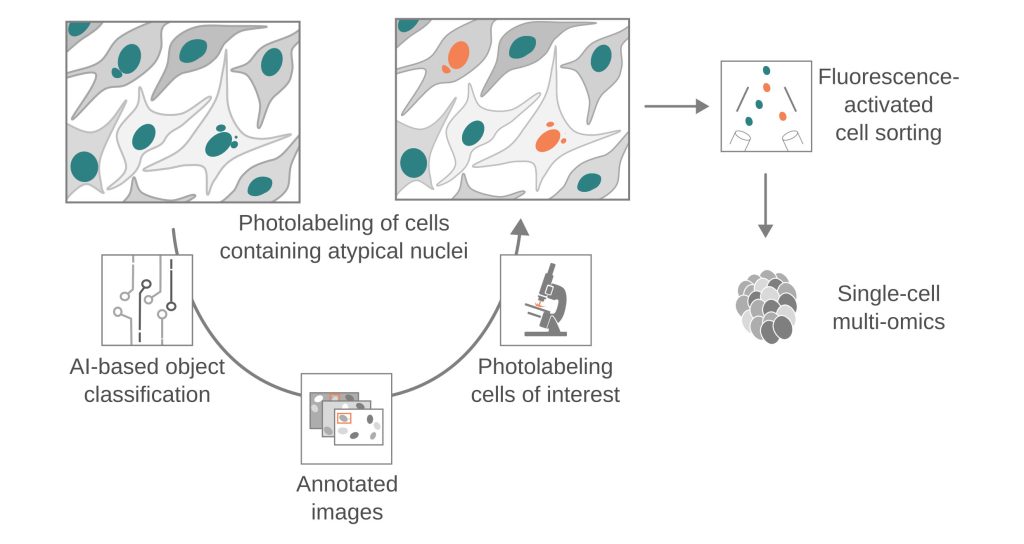
To dissect these complex phenomena, we leverage and integrate innovative technologies, which include long-read sequencing, multi-omics techniques, adaptive-feedback microscopy, and data science methodology. These approaches enable us to study genomic alterations at an unprecedented scale and resolution, providing insights into mechanisms of chromosomal instability and cellular heterogeneity. Our MAGIC platform, for example, couples automated live-cell imaging and single-cell multi-omics with machine learning to systematically explore de novo chromosomal rearrangements, revealing biological pathways that govern chromosome stability in human cells.
Key recent contributions
- Chromosomal instability: We have developed MAGIC (Machine-learning Assisted Genomics-and-Imaging Convergence), which integrates on-the-fly imaging and single-cell multi-omics to investigate the basal rates and molecular mechanisms of de novo chromosomal rearrangements, enhancing our understanding of chromosomal instability in both non-malignant and malignant contexts.
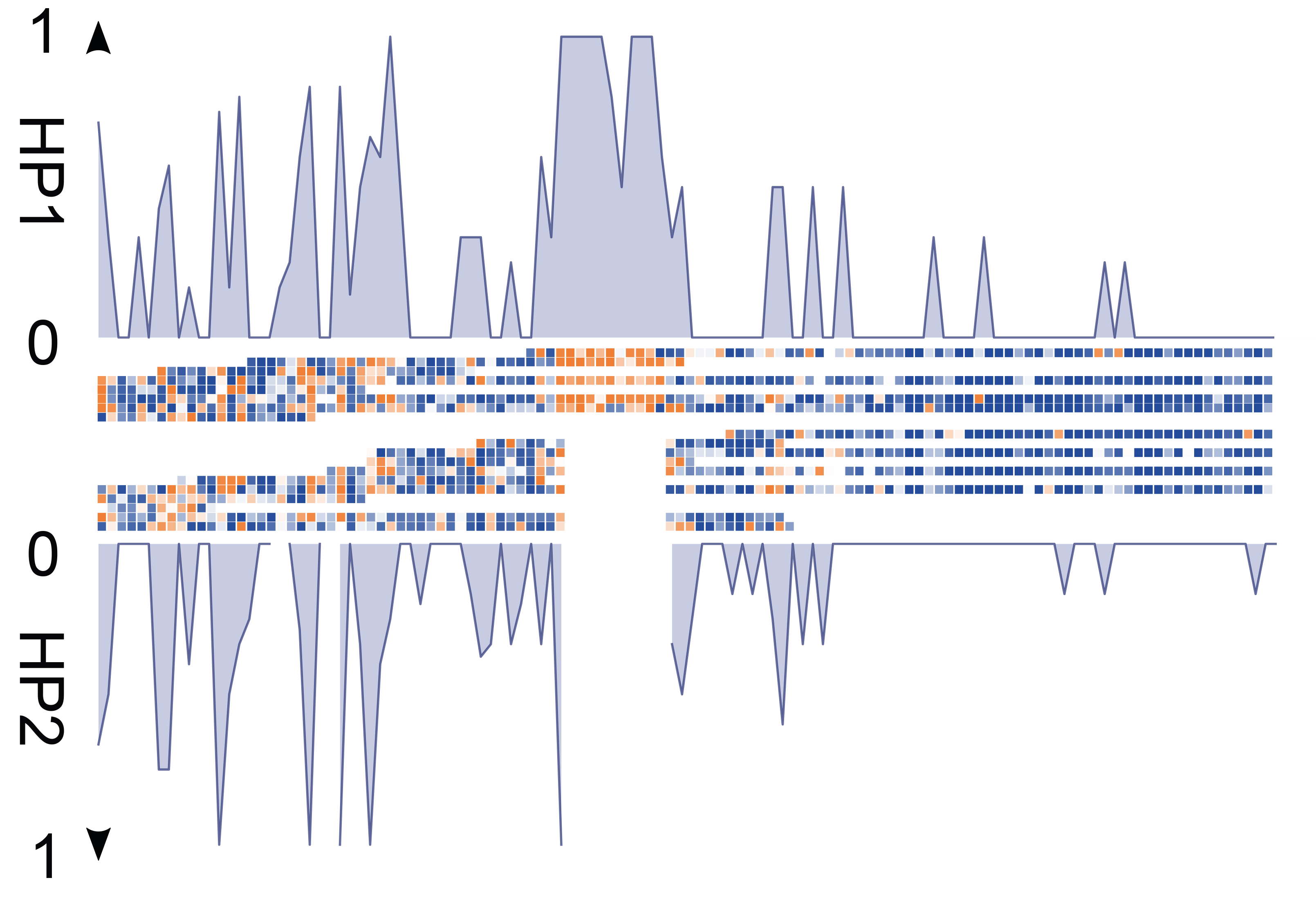
- Somatic SV mosaicisms: Using Strand-seq, we have characterized the extent of SV mosaicism in hematopoietic stem and progenitor cells, revealing the enrichment of SVs in specific cell types with age, and their role in dysregulating cellular pathways.
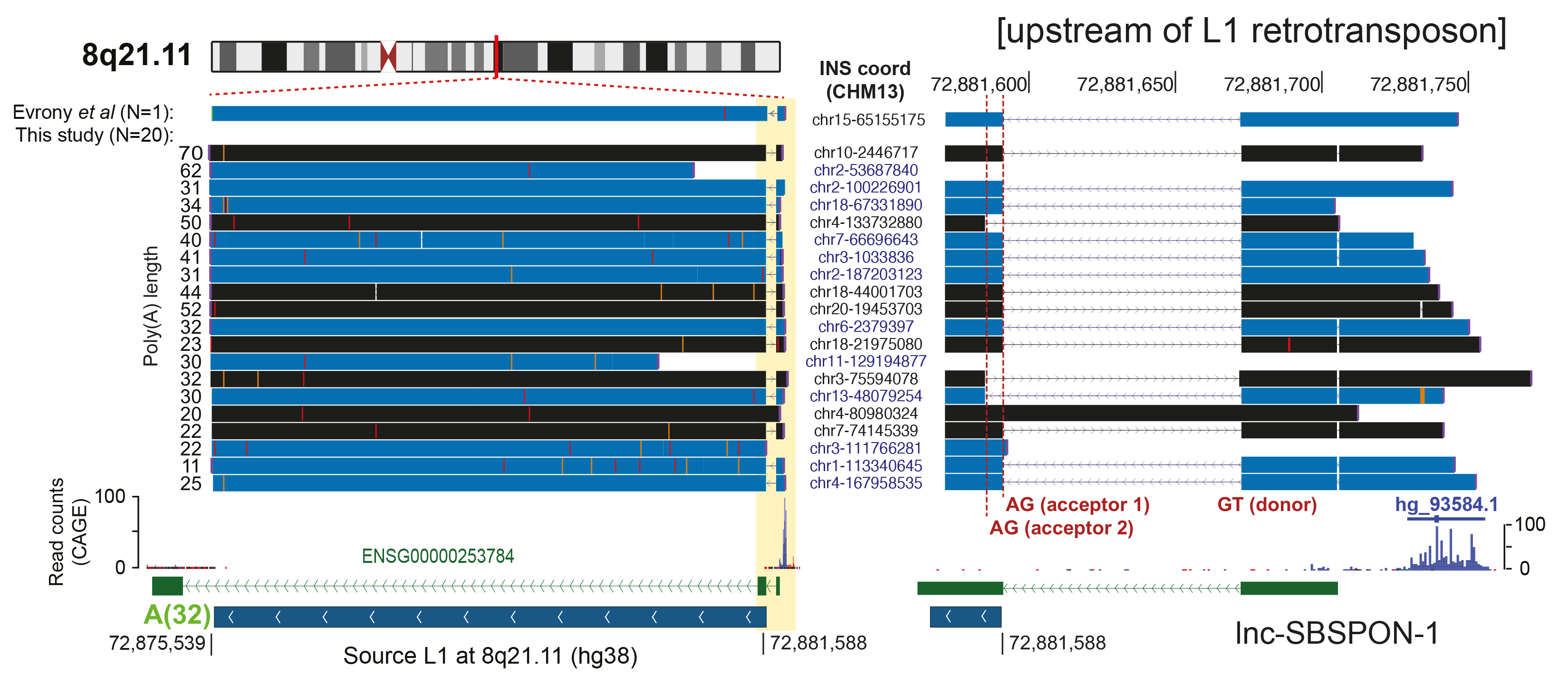
- Population-scale human genome assembly and SV analysis: Using long-read sequencing, we constructed comprehensive population-scale SV catalogs from 1,019 genomes from the 1000 Genomes Project and 65 near telomere-to-telomere assemblies. These resources revealed new insights into SV formation mechanisms and functional impacts, such as the role of retrotransposons in mediating genetic variation.
- Mechanisms of cancer development: Our research into cancer genomes has uncovered unique mutational processes in brain tumors and acute leukemia, such as a “three-hit” model involving germline mutations and somatic chromosomal rearrangements revealing mechanistic insights into the tumorigenesis of medulloblastoma.
Future directions
- Chromosomal rearrangement processes: Dissecting mitosis-driven genome instability processes using advanced imaging and single-cell sequencing techniques coupled with machine learning.
- Population diversity and functional consequences of genome variation: Expanding genetic variation maps with long-read sequencing to explore population-scale genetic diversity and its clinical implications.
- Somatic 3D genomics: Integrating spatial and single-cell omics with bioimaging to elucidate principles of cellular and genetic heterogeneity in human tissues.
- Multi-omics data integration: Linking genetic and epigenetic mosaicism to uncover their impact on complex phenotypes including cancer.
ERC INVESTIGATOR