Human genomes provide new reference for global genetic diversity
EMBL scientists co-initiated the reconstruction of the most diverse set of reference human genomes ever assembled
Credit: David Porubsky/University of Washington
In 2001, the International Human Genome Sequencing Consortium announced the first draft of the human genome reference sequence. The Human Genome Project had taken more than 11 years of work and involved more than 1,000 scientists from 40 countries. The reference sequence, however, does not represent a single individual but instead is a composite of genomes from several individuals that cannot accurately capture the complexity of human genetic variation.
Building on this, scientists have carried out many sequencing projects over the past 20 years to identify and catalogue genetic differences between individual genomes and the reference genome. These differences usually focused on small changes of a single letter of the DNA code and missed larger genetic alterations. Current technologies are now beginning to detect and characterise larger differences – called structural variants – such as insertions of several hundred letters. Structural variants are more likely than smaller genetic differences to interfere with gene function.
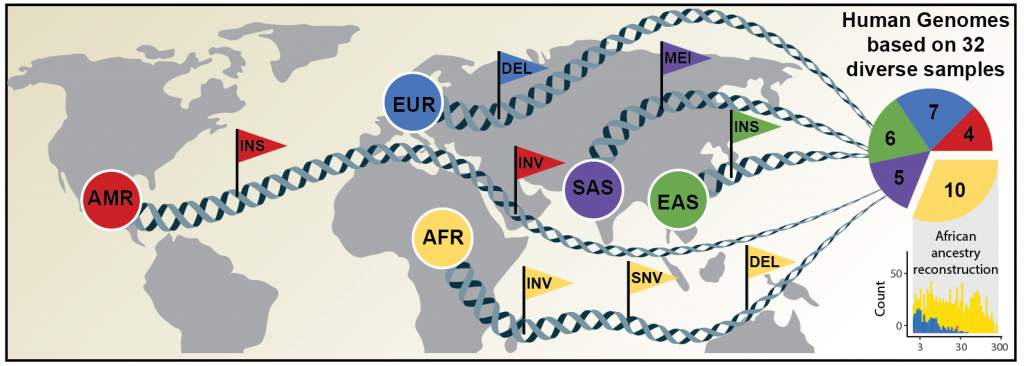
EMBL’s Korbel group, in partnership with Heinrich Heine University Düsseldorf, The Jackson Laboratory in Farmington, Connecticut, and the University of Washington in Seattle, has now published an article in Science announcing a new, significantly more comprehensive reference dataset obtained using a combination of advanced sequencing and mapping technologies. The new reference dataset contains 64 assembled human genomes, representing 25 human populations from Africa, North America, East and South Asia, and Europe.
This study builds on a new method published by the researchers last year in Nature Biotechnology to accurately reconstruct the two components of a person’s genome – one inherited from each of their parents. When assembling a person’s genome, this method relied on a technology provided by EMBL known as Strand-seq to distinguish maternal and paternal DNA sequences.
“For each human individual that participated in the study, we identified not one but two genomes – one for each set of chromosomes,” explains Jan Korbel, who led the research at EMBL. “Humans have two sets of chromosomes, which they receive from their parents. Previously we could not distinguish whether genetic variation came from one chromosome set or the other. We have now been able to solve this thanks to advances made by the Human Genome Structural Variation Consortium. It represents a remarkable achievement for the discovery of genetic variation in humans, which can now be studied much more comprehensively, leading the way to better find disease-causing genes.”
The distribution of genetic variants can differ substantially between population groups as a result of spontaneous and continuously occurring changes in the genetic material. If such a mutation is passed on over many generations, it can become a genetic variant specific to that population.
The new reference data provide an important basis for including the full spectrum of genetic variants in genome-wide association studies, which examine genetic variants across the whole genome to find out whether any variants are associated with specific traits or diseases. The aim is to estimate an individual’s risk of developing diseases such as cancer, and to understand the underlying molecular mechanisms. This, in turn, can be used as a basis for more targeted therapies and preventive medicine.
This work might enable further applications in precision medicine. Drug efficacy, for example, can vary between individuals based on their genomes. The new reference data now represent the full range of genetic variant types and incorporate human genomes of great diversity. “These genomes will enable a new wave of scientific discoveries about the biology of the human genome and the connection between genetic variation and disease,” says EMBL researcher and co-first author Bernardo Rodriguez-Martin. “As an example, we were able to estimate the age of highly mutagenic L1 repeats. Very surprisingly, although these sequences originated up to three million years ago, they continue to mutate the human genome frequently, which occasionally leads to diseases such as cancer.” This new resource might therefore contribute to developing novel approaches in personalised medicine, where the selection of therapies is tailored to a patient’s individual genetic background.
Menschliche Genome als neue Referenz für die globale genetische Vielfalt
EMBL-Wissenschaftler mitinitiieren Erstellung des vielfältigsten Satzes menschlicher Referenzgenome, der jemals zusammengestellt wurde
Im Jahr 2001 gab das International Human Genome Sequencing Consortiumden ersten Entwurf der Referenzsequenz des menschlichen Genoms bekannt. Das Humangenomprojekt, wie es genannt wurde, hatte mehr als elf Jahre Arbeit in Anspruch genommen und mehr als 1000 Wissenschaftler aus 40 Ländern beteiligt. Diese Referenz repräsentierte jedoch kein einzelnes Individuum, sondern ein Gemisch von Menschen, das die Komplexität der menschlichen genetischen Variation nicht genau erfassen konnte.
Darauf aufbauend haben Wissenschaftler in den letzten 20 Jahren viele Sequenzierprojekte durchgeführt, um genetische Unterschiede zwischen individuellen Genomen und dem Referenzgenom zu identifizieren und zu katalogisieren. Diese Unterschiede konzentrierten sich in der Regel auf Änderungen einzelner Buchstaben des DNA-Codes und ließen größere genetische Veränderungen außer Acht. Moderne Technologien beginnen nun, größere Unterschiede – sogenannte strukturelle Varianten – zu erkennen und zu charakterisieren. Strukturelle Varianten sind wahrscheinlicher als kleinere genetische Unterschiede, beteiligt wenn Genfunktionen durch Mutationen beeinträchtigt werden.
Die Korbel-Gruppe am EMBL hat nun in Zusammenarbeit mit der Heinrich-Heine-Universität Düsseldorf, dem Jackson Laboratory in Farmington, Connecticut, und der Universität Washington in Seattle einen Artikel in der Zeitschrift Science veröffentlicht, in dem sie einen neuen, wesentlich umfangreicheren Referenzdatensatz vorstellen, der mit einer Kombination aus fortschrittlichen Sequenzier- und Kartierungstechnologien gewonnen wurde. Der neue Referenzdatensatz enthält 64 menschliche Genome, die 25 menschliche Populationen aus Afrika, Nordamerika, Ost- und Südasien und Europa repräsentieren.
Diese Studie baut auf einer neuen Methode auf, die von der Forschungsgruppe letztes Jahr in Nature Biotechnology veröffentlicht wurde. Mit dieser lassen sich die beiden Komponenten des Genoms – die väterliche und die mütterliche – einer Person genau rekonstruieren. Beim Zusammensetzen des Genoms einer Person nutzt diese Methode eine vom EMBL bereitgestellte Technologie, das sogenannte Strand-seq, um die beiden DNA-Sequenzen zu unterscheiden.
„Für jedes menschliche Individuum, das an der Studie teilgenommen hat, haben wir nicht ein, sondern zwei Genome identifiziert – eines für jeden Chromosomensatz,“ erklärt Jan Korbel, der die Forschung am EMBL geleitet hat. „Der Mensch hat zwei Chromosomensätze, die er von seinen Eltern erhält. Bisher konnten wir nicht unterscheiden, ob die genetische Variation von dem einen oder dem anderen Chromosomensatz stammt. Dies konnten wir nun dank der Fortschritte des Konsortiums lösen. Das ist ein bemerkenswerter Erfolg für die Entdeckung genetischer Variationen beim Menschen, die nun viel umfassender untersucht werden können, was wiederum den Weg zu einer besseren Suche nach krankheitsverursachenden Genen ebnet.“
Die Verteilung der genetischen Varianten kann sich durch spontane und kontinuierlich auftretende Veränderungen im Erbgut zwischen Bevölkerungsgruppen erheblich unterscheiden. Wird eine solche Mutation über viele Generationen weitergegeben, kann sie zu einer für diese Population spezifischen genetischen Variante werden.
Die neuen Referenzdaten bilden eine wichtige Grundlage für die Einbeziehung des gesamten Spektrums genetischer Varianten in genomweite Assoziationsstudien. Bei diesen werden genetische Varianten über das gesamte Genom hinweg untersucht, um herauszufinden, ob bestimmte Varianten mit bestimmten Merkmalen oder Krankheiten assoziiert sind. Ziel ist es, das individuelle Risiko für die Entstehung von Krankheiten – wie etwa Krebs – abzuschätzen und die zugrunde liegenden molekularen Mechanismen zu verstehen. Dies wiederum kann als Grundlage für gezieltere Therapien und Präventivmedizin genutzt werden.
„Diese Genome werden den Weg für eine neue Welle wissenschaftlicher Entdeckungen über die Biologie des menschlichen Genoms und den Zusammenhang zwischen genetischer Variation und Krankheit ebnen,“ sagt EMBL-Forscher und Co-Erstautor Bernardo Rodriguez-Martin. Er fügt hinzu: „Wir konnten zum Beispiel das Alter von hoch mutagenen L1-Elementen im Erbgut abschätzen. Sehr überraschend ist, dass diese Sequenzen, obwohl sie vor bis zu drei Millionen Jahren entstanden sind, immer noch häufig im menschlichen Genom mutieren, was gelegentlich zu Krankheiten wie Krebs führt.“ So könnte diese neue Ressource dazu beitragen, neue Ansätze in der personalisierten Medizin zu entwickeln, bei der die Auswahl von Therapien auf den individuellen genetischen Hintergrund eines Patienten zugeschnitten wird.
Nuevos genomas proveen una referencia para la diversidad genética humana
Científicos del EMBL, junto con otros investigadores, han empezado la reconstrucción del conjunto más diverso de genomas de referencia jamás reunido
En 2001, el Consorcio Internacional de Secuenciación del Genoma Humano anunció el primer borrador de la secuencia de referencia del genoma humano. El Proyecto Genoma Humano había llevado más de once años de trabajo e involucró a más de 1000 científicos de 40 países. Esta secuencia de referencia, sin embargo, no representa a una sola persona, sino que es una combinación de humanos, que no puede capturar con precisión la complejidad de la variación genética humana.
Sobre esta base, durante los últimos 20 años, se han llevado a cabo muchos proyectos de secuenciación para identificar y catalogar las diferencias genéticas entre un individuo y el genoma de referencia. Esas diferencias generalmente consistían en pequeños cambios de una sola base y pasaban por alto alteraciones genéticas más grandes. Las tecnologías actuales ahora pueden comenzar a detectar y caracterizar diferencias más significativas, llamadas variantes estructurales, como inserciones de varios cientos de letras. Las variantes estructurales tienen más probabilidades de interferir con la función genética que las diferencias genéticas más pequeñas.
El grupo de investigación del EMBL liderado por Jan Korbel, en asociación con la Universidad Heinrich Heine de Düsseldorf, el Laboratorio Jackson en Farmington, Connecticut, y la Universidad de Washington en Seattle, ha publicado un artículo en la revista Science en el que ha anunciado un nuevo conjunto de datos de referencia mucho más completo obtenido utilizando una combinación de tecnologías avanzadas de secuenciación y mapeo. El nuevo conjunto de datos de referencia consiste en 64 genomas humanos ensamblados, que representan 25 poblaciones humanas diferentes de África, América del Norte, Asia Oriental y Meridional, y Europa.
Este estudio se basa en un nuevo método publicado por estos investigadores el año pasado en la revista Nature Biotechnology para reconstruir con precisión los dos componentes del genoma de una persona: el heredado por cada uno de los progenitores. Al ensamblar el genoma de una persona, este método utiliza una tecnología desarrollada por el EMBL (denominada Strand-seq) para distinguir las secuencias de ADN paternas y maternas.
“Para cada persona que ha participado en el estudio, identificamos no uno, sino dos genomas, uno para cada conjunto de cromosomas”, explica Jan Korbel, quien dirigió la investigación en el EMBL. “Los seres humanos tenemos dos conjuntos de los cromosomas que recibimos de nuestros padres. Anteriormente, no podíamos distinguir si la variación genética provenía de los cromosomas del uno o del otro. En cambio, gracias a los avances realizados por el Consorcio de Variación Estructural del Genoma Humano, ahora sí hemos sido capaces de hacerlo. Se trata de un logro extraordinario para estudiar la variación genética en humanos, que ahora se puede estudiar de manera mucho más completa, lo que abre el camino para encontrar genes causantes de enfermedades”.
La distribución de variantes genéticas puede diferir sustancialmente entre grupos de población como resultado de cambios espontáneos y que ocurren continuamente en el material genético. Si dicha mutación se transmite a lo largo de muchas generaciones, puede convertirse en una variante genética específica de esa población.
Los nuevos datos de referencia proporcionan una base importante para incluir en los denominados estudios de asociación de todo el genoma el espectro completo de variantes genéticas. El objetivo es estimar el riesgo individual de desarrollar ciertas enfermedades como el cáncer y comprender los mecanismos moleculares subyacentes. Esto, a su vez, puede usarse como base para terapias más dirigidas y medicina preventiva.
Este trabajo podría permitir nuevas aplicaciones en la medicina de precisión. La eficacia de un fármaco, por ejemplo, puede variar entre individuos en función de sus genomas. Los nuevos datos de referencia ahora representan la gama completa de diferentes tipos de variantes genéticas e incorporan genomas humanos de gran diversidad. “Estos genomas abrirán el camino hacia una nueva ola de descubrimientos científicos sobre la biología del genoma humano y la conexión entre variación genética y enfermedades”, dice el investigador del EMBL y co-primer autor Bernardo Rodríguez-Martín. “Como ejemplo, hemos podido estimar la edad de las repeticiones altamente mutagénicas de L1. Sorprendentemente, aunque estas secuencias se originaron hace un máximo de tres millones de años, continúan mutando el genoma humano con frecuencia, cosa que ocasionalmente conduce a enfermedades como el cáncer”. Este nuevo recurso podría contribuir al desarrollo de enfoques novedosos en la medicina personalizada, en la que la selección de terapias se adapta a los antecedentes genéticos individuales de cada paciente.