EMBL-EBI Press Office
Wellcome Genome Campus Hinxton, Cambridgeshire CB10 1SD, UK
media@ebi.ac.uk+44 1223 494369
A new computational tool allows the most accurate insights into evolution ever
What makes a human different from a chimp? Researchers from the European Molecular Biology Laboratory’s European Bioinformatics Institute (EMBL-EBI) have come one important step closer to answering such evolutionary questions correctly. In the current issue of Science they uncover systematic errors in existing methods that compare genetic sequences of different species to learn about their evolutionary relationships. They present a new computational tool that avoids these errors and provides accurate insights into the evolution of DNA and protein sequences. The results challenge our understanding of how evolution happens and suggest that sequence turnover is much more common than assumed.
“Evolution is happening so slowly that we cannot study it by simply watching it. That’s why we learn about the relationships between species and the course and mechanism of evolution by comparing genetic sequences,” says Nick Goldman, group leader at EMBL-EBI.
The four letter code that constitutes the DNA of all living things changes over time; for example individual or several letters can be copied incorrectly (substitution), lost (deletion) or gained (insertion). Such changes can lead to functional and structural changes in genes and proteins and ultimately to the formation of new species. Reconstructing the history of these mutation events reveals the course of evolution.
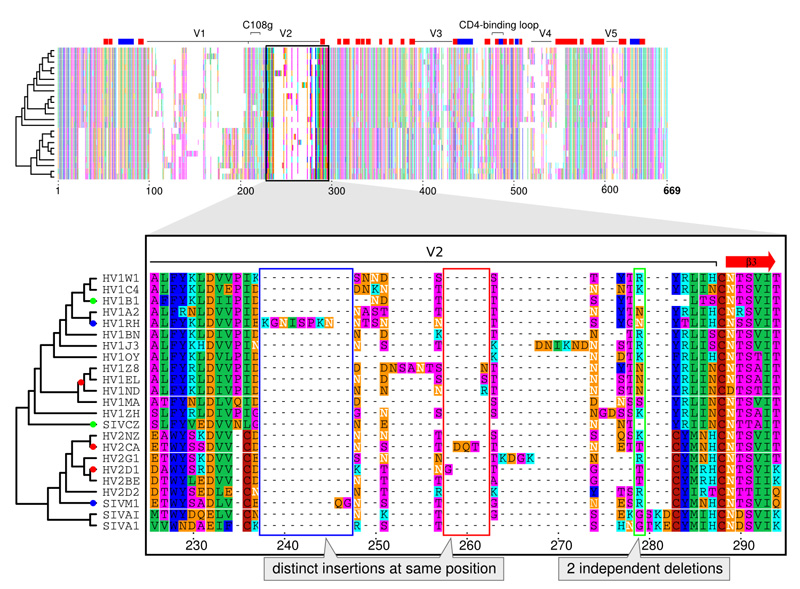
A comparison of multiple sequences starts with their alignment. Characters in different sequences that share common ancestry are matched and gains and losses of characters are marked as gaps. Since this procedure is computationally heavy, multiple alignments are often built progressively from several pairwise alignments. It is impossible, however, to judge if a length difference between two sequences is a deletion in one or an insertion in the other sequence. For correct alignment of multiple sequences, distinguishing between these two events is crucial. Existing methods, that fail to do that, lead to a flawed understanding of the course of evolution.
“Our new method gets around these errors by taking into account what we already know about evolutionary relationships,” says Ari Löytynoja, who developed the tool in Goldman’s lab. “Say we are comparing the DNA of human and chimp and can’t tell if a deletion or an insertion happened. To solve this our tool automatically invokes information about the corresponding sequences in closely related species, such as gorilla or macaque. If they show the same gap as the chimp, this suggests an insertion in humans.”
Findings achieved with the new technique suggest that insertions are much more common than assumed, while the frequency of deletions has been overestimated by existing methods. A likely reason for these systematic errors of other techniques is that they were originally developed for structural matching of protein sequences. The focus of molecular biology is shifting, however, and understanding functional changes in genomes requires specifically designed methods that consider sequences’ histories. Such approaches will likely reveal further bugs in our understanding of evolution in future and might challenge the conventional picture of sequence evolution.